- Research article
- Open access
- Published:
Structural model of nicotinic acetylcholine receptor isotypes bound to acetylcholine and nicotine
BMC Structural Biology volume 2, Article number: 1 (2002)
Abstract
Background
Nicotine is a psychoactive drug presenting a diverse array of biological activities, some positive, such as enhancement of cognitive performances, others negative, such as addiction liability. Ligands that discriminate between the different isotypes of nicotinic acetylcholine receptors (nAChRs) could present improved pharmacology and toxicity profile.
Results
Based on the recent crystal structure of a soluble acetylcholine binding protein from snails, we have built atomic models of acetylcholine and nicotine bound to the pocket of four different human nAChR subtypes. The structures of the docked ligands correlate with available biochemical data, and reveal that the determinants for isotype selectivity are relying essentially on four residues, providing diversity of the ligand binding pocket both in terms of Van der Waals boundary, and electrostatic potential. We used our models to screen in silico a large compound database and identify a new ligand candidate that could display subtype selectivity.
Conclusion
The nAChR-agonist models should be useful for the design of nAChR agonists with diverse specificity profiles.
Background
The nicotinic acetylcholine receptor (nAChR) is a ligand-gated ion channel that mediates neurotransmission at the neuromuscular junction, autonomic ganglia and at some sites in the central nervous system. Distinct nAChR subtypes exist that can be stimulated by the neurotransmitter acetylcholine, the natural product nicotine, or by synthetic compounds [1]. nAChR is the prototype for a protein superfamily that includes the receptors for the excitatory amino acids glutamate and aspartate, the inhibitory amino-acids gamma-aminobutyric acid (GABA) and glycine, as well as the serotonin 5-HT3 receptor [1]. Like most ligand-gated ion channels, nAChRs are homo- or hetero-pentameric, each monomer belonging to a pull of 8 alpha- or 3 beta-subunits (alpha2-9, beta2-4). While each subunit has four transmembrane domains, the agonist binding sites are located at the subunit interfaces [2]. Both natural and synthetic ligands can display varied affinity for different receptor subtypes. For instance, nicotine has a Kd of 1 nM for alpha4-beta2 nAChR, but only 4 μM for the alpha7-alpha7 isoform [3].
In recent years, it has become clear that the neuronal nAChR is a valid target against a variety of diseases, including cognitive and attention deficits, Parkinson's disease, anxiety, and pain management [3, 4]; the addiction liability and other undesirable side-effects of nicotine prohibit the use of this natural product for therapeutic applications, but the active development of nAChR agonists presenting adequate receptor subtype specificity should result in improved pharmacology and potency [4]. As for most membrane proteins, information on the 3 dimensional structure of the nAChR is limited, and the best experimental structure consists of 4.6 A data from electron microscopy [5], a resolution that does not allow for structure-based design approaches.
Recently, Sixma and colleagues have determined the first high-resolution structure of a soluble acetylcholine binding protein (AChBP), extracted from snail neurons, and homologous to the ligand binding domain of nAChR [6]. Based on this high-resolution crystal structure, we have built an atomic model of the agonist binding site of nAChR, and docked acetylcholine and nicotine to different receptor isotypes. The resulting models provide a valuable framework for structure-based design of subtype-specific nAChR agonists.
Results and Discussion
The crystal structure of two consecutive AChBP monomers [6] was used as a template to build a homology-model of four known native human nAChR subunit interfaces: alpha4-beta2 (I1), alpha4-beta4 (I2), alpha3-beta2 (I3), and alpha7-alpha7 (I4). The corresponding first two nAChR subtypes are possibly involved in cognition, neurodegeneration, pain, anxiety and depression, while alpha3-beta2 nAChR is associated with dopamine release and addiction, and alpha7-alpha7 with GABA release [4].
First, the sequences of the five nAChR monomers (alpha3, alpha4, alpha7, beta2 and beta4) were aligned to AChBP in a multi-protein sequence alignment. The alignment was generated automatically by the ICM method and adjusted manually around residues 188–208, to align an insertion in human nAChR subunits with a loop region of the 3D template (Figure 1).

Multiple sequence alignment of AChBP with nAChR monomers. The alignment was divided into subunit 1 monomers (alpha3, alpha4, alpha7) (top) and subunit 2 monomers (beta2, beta4, and alpha7) (bottom), in order to preserve separate residue numbering consistent with human alpha4 and beta2 nAChR. Side-chain residues contacting docked acetylcholine and nicotine are colored gold in subunit 1 and cyan in subunit 2. Additional residues forming the ligand binding pocket boundary but further from the ligands are shown in bold. Non-conserved residues that can be used to design isotype selective agonists are underlined, and marked by a "!" in the numbering line. AChBP residues contacting HEPES in the crystal structure of the complex [6] have a shaded background. Secondary structure elements are shown under the sequences : H=alpha helix, E = beta-strand, B=beta-bridge.
Based on the sequence alignment, the query sequences where threaded onto the first two subunits of the AChBP pentamer. A disulfide bridge (the so-called cys-loop), present only in the alpha subunits of nAChR, is known to be part of the agonist binding site of the receptor [7]. This was a clear indication that, in order to build a dimer interface that includes the ligand binding pocket, alpha subunits had to be aligned onto the template monomer presenting a disulfide bridge at the interface (subunit 1). A putative HEPES (N-2 hydroxyethylpiperazine-N'-2-ethanesulphonic acid) molecule is present in the binding pocket of the template structure. All residues in the close vicinity of this ligand were confirmed experimentally as being part of the acetylcholine binding pocket [6]. While the homology between AChBP and the nAChR subunits is relatively low (22 % sequence identity, for instance, between alpha4 and AChBP), and the overall model needs to be refined, 72% of the AChBP residues in contact with HEPES are identical in the alpha4-beta2 nAChR (Figure 1), and the predicted conformation of the agonist binding pocket of nAChR is expected to be accurate. In order to optimize the conformation of this pocket, all residues within 7A of the HEPES molecule that were not identical between template and model were energy-minimized in the internal coordinate space [8]. The resulting models were used for docking experiments.
Both acetylcholine and nicotine were docked to our model of the alpha4-beta2 dimer interface. A formal charge of +1 was assigned to the quaternary ammonium group of acetylcholine and protonated nicotine. Indeed, a correlation between EC50 values and quantum mechanics simulations strongly suggests the existence of a cation-pi interaction between a triptophan residue of the ligand binding pocket and the quaternary ammonium of acetylcholine [9]. A series of grid representations of the ligand binding pocket was generated that accounted for Van der Waals shape, electrostatic potential, as well as hydrogen bonding and hydrophobicity profiles. The flexible ligands were rapidly docked into the superimposed grids, and the lowest energy conformation was further refined with an atomic representation of the receptor, and with both ligand and receptor side chains flexible [10] (see "Materials and Methods" for details).
Acetylcholine fits tightly into the agonist binding site at the alpha4-beta2 interface of nAChR (Figure 2). This binding site coincides with that of AChBP, and is in agreement with photo-affinity labeling and mutagenesis study [6, 11]. The ligand makes Van der Waals contact with Trp182, Tyr230 and Cys225-226 of alpha4, and Trp82, Leu146 and Phe144 of beta2 (Figure 3 – Top). The charged ammonium nitrogen is closest to the side-chain of alpha4 Trp182. This is in agreement with previous work assigning a cation-pi interaction between acetylcholine and this residue exclusively [9]. The carbonyl oxygen of acetylcholine and backbone nitrogen of Leul46 are also engaged in a hydrogen bond. Interestingly, a small polar cleft next to this oxygen could accommodate a water molecule coordinated by two hydrogen bond donors -alpha4 Trp182 indole nitrogen and beta2 Leu146 amide nitrogen- and one hydrogen bond acceptor -beta2 Ser133 carbonyl (Figure 3 – Left). This water molecule would form an additional hydrogen bond with acetylcholine, thereby stabilizing further the predicted bound conformation. Surprisingly, the ester moiety of acetylcholine does not seem to interact with the cys-loop of the receptor, as previously suggested by photoaffinity labeling [12], but is engaged in similar type of interaction as that observed with the acidic group of HEPES in the AChBP crystal structure [6]. It is possible that the reactive di-azo group of the photosensitive agonist used in photoaffinity labeling would extend towards the cys-loop, while the conformation of the bound agonist remains in agreement with our model.

Acetylcholine (space filling representation) docked into the agonist binding pocket of human nAChR, at the interface of the alpha4 (subunit 1) and beta2 (subunit 2) monomers. Gold: alpha4, cyan: beta2, yellow: ligand binding site. Other interfaces modeled in this study are also indicated.

Acetylcholine (top) and nicotine (bottom) docked to the agonist binding site of human nAChR alpha4-beta2. Left: the ligands would form a polar interaction with the delocalized electron of Trp182, and with a water molecule (green) coordinated by Trp182, Leu146 and Ser133. Right: space filling representation shows how the ligands occupy the binding pocket (yellow). Color coding is magenta: ligand, gold: subunit alpha4, cyan: subunit beta2, blue: nitrogen, red: oxygen, yellow: sulfur. Most hydrogen atoms (gray) are not shown for clarity.
Nicotine makes Van Der Waals contacts with the same array of receptor side-chains (Figure 3 – Bottom). The protonated ammonium nitrogen is also making polar interaction with the delocalized electron and backbone carbonyl of Trp182, and is less than 1A away from the quaternary nitrogen of docked acetylcholine. Additionally, a hydrogen bond between the pyridine nitrogen of nicotine and indole nitrogen of Trp182 is possible (the two atoms are 3.4 A apart), but the bond angle is not optimal (70 degrees with the plane of the aromatic side-chain). Interestingly, while two conformations appear with equal probability when the docking is performed without the water molecule positioned in the acetylcholine complex, addition of this water molecule results in an extra hydrogen bond with the pyridine nitrogen of nicotine, and stabilizes the conformation presented (Figure 3 – Bottom). In the alternative, less energetically favorable conformation, nicotine contacts mostly subunit alpha4, plus Leu146 and Trp82 of beta2 (data not shown).
Similarly, acetylcholine and nicotine were docked into the agonist binding site at the alpha3-beta2, alpha4-beta4 and alpha7-alpha7 interfaces. The ligands adopted similar conformation as docked to alpha4-beta2. Four residues in the vicinity of the ligands are varying from one receptor isotype to the other: Thr183 of alpha4 (Ser in alpha3 and alpha7), Ala135 of beta2 (Leu in beta4 and Val in alpha7), Phe144 of beta2 (Leu in beta4 and Gln in alpha7), and Thr83 of beta2 (Lys in beta4 and Gln in alpha7) (Figure 1). Neither a threonine, nor a serine at position 183 of subunit 1 contact the ligands. On the other hand, while Ala135 is 8.5A away from the docked agonists, a leucine could make some hydrophobic interaction with nicotine and acetylcholine. The side-chain of Phe144 contacts the ligands only via its c-alpha an c-beta atoms, and similar interaction can occur with a leucine or glutamine at this position; however, while the conformation presented is projecting away from the ligands, alternative conformers could bring the side-chain closer to the agonists, and represent structural determinants for specificity. At last, beta-2 Thr83 is rather remote from the ligands (8.5 A), but the side-chain of a lysine could come closer and influence lightly the electrostatic potential of the binding cavity.
Based on our models, we calculated the binding energy of acetylcholine and nicotine to the four receptor isotypes in the presence of the water molecule (Table 1 – top). The binding energy function, previously validated against a diversified benchmark [20], includes a hydrophobic term, an electrostatic term, and an entropic term, but does not include Van der Waals energy, since this term is too noisy; Van der Waals interactions are supposed to be proportional to the interface of the interacting molecules, and incorporated into the surface (hydrophobic) term (see Materials and Methods). As a result, the binding energy function has an accuracy of 2.5 kcal/mol, but does not account for potential Van der Waals clashes. The Van der Waals interaction energies were calculated separately, and are also presented.
Table 1 shows that binding energy differences between the two ligands and the four nAChR subtypes are rather small, and within the 2.5 kcal/mol accuracy of the estimates [20]. As expected, no Van der Waals clash is observed. The calculated energies suggest a lower affinity of nicotine for alpha7-alpha7 than for alpha4-beta2, which is in agreement with experiments [3]. The ligands were also docked to the crystal structure of AChBP (the binding conformation was identical as with nAChRs), and the binding energies calculated. Again, the predicted energies of acetylcholine and nicotine binding to AChBP are close to those for binding to nAChRs (Table 1 – top). We also docked HEPES to the receptors, since this ligand was found in the crystal structure of AChBP. Low-energy conformations were found only in the absence of water molecule, and the calculated binding energies (Table 1 – bottom) suggest that HEPES is a poor ligand for nAChRs, and AChBP, and was observed in the crystal structure because of its high concentration (100 mM) in the crystallization condition [6].
The presence of the isotype diversity of nAChR ligand binding pocket should allow for the rational design of selective agonists. Variations at residues alpha4 Thr183 and beta2 Ala135 generate different shapes of the binding pocket (Figure 4 – Top), but preserve the electrostatic potential of the system: all non-conserved residues at position 183 are hydrophobic, and hydroxyl of Thr154 and Ser154 are superimposed, pointing away from the ligand, towards the carboxylate of Asp122. On the other hand, variations at residues 83 and 144 of subunit 2 can modify lightly the charge distribution of the binding site. Coloring of the pocket according to its electrostatic potential shows for instance that the alpha7-alpha7 pocket is slightly more electronegative than alpha4-beta2 (Figure 4 – Bottom). However, these variations are rather limited and the agonist binding site remains electronegative for all isotypes tested.

Structural determinants for receptor subtype specificity. Top: Two non-conserved residues provide structural diversity to the ligand binding site (left: alpha4-beta2, center: alpha4-beta4, right: alpha7-alpha7). The shape difference of the Van der Waals boundary is shown at residue 183 of subunit 1 (gold), and 135 of subunit 2 (cyan), next to docked nicotine (magenta). Bottom: Electrostatic potential coloring shows a rather limited variation in charge distribution between the alpha4-beta2 dimer (left) and the alpha7-alpha7 dimer (right). Charge color coding is red: negative, gray: hydrophobic, blue: positive.
In order to illustrate how, based on our models, a high affinity ligand can be rationally designed to be specific for beta2-containing nAChR subtypes, we searched a database of over 200,000 compounds (ChemDiv Inc., San Diego, CA), for alpha4-beta2 ligand candidates presenting a functional group close to Ala 135 of the beta2 subunit, hoping that such functional group could not accommodate the bulkier Leu 135 of beta4 (Figure 4 – Top). Each compound of the database was docked in the grid potential representation of the alpha4-beta2 receptor (without a water molecule), and assigned a score reflecting the quality of the fit, as implemented in the ICM-VLS module (Molsoft LLC, San Diego, CA). The top scoring compounds were considered as good ligand candidates and were screened with two additional filters.
-
1)
They should present a functional group within 5 A of beta2 Ala135. 2) They should have a polar group that fills the cavity occupied by water in the nicotine and acetylcholine complexes, and that makes hydrogen bonds similar to the water molecule (Figure 3 – Left). We believe this second filter should result in the identification of improved pharmacophores for nAChR ligands.
Molecule 1 is one good candidate that matches all criteria (Figure 5A).
-
1)
It is the 16th best scoring compound, and displays better calculated binding energies than acetylcholine or nicotine for alpha4-beta2 nAChR (Table 1 – Bottom).
-
2)
It presents a pyridine in the vicinity of beta2-Ala135, which clashes with the corresponding Leu 135 of beta4 (Figures 5B and 5D).
-
3)
It displays an amide group that occupies a cavity not filled by nicotine or acetylcholine, and makes a network of hydrogen bonds with the indole nitrogen of alpha4-Trp182, the backbone carbonyl of beta2-Ser133 and beta2-Phe144, and with the backbone nitrogen of beta2-Leu146 (Figure 5B).
Figure 5 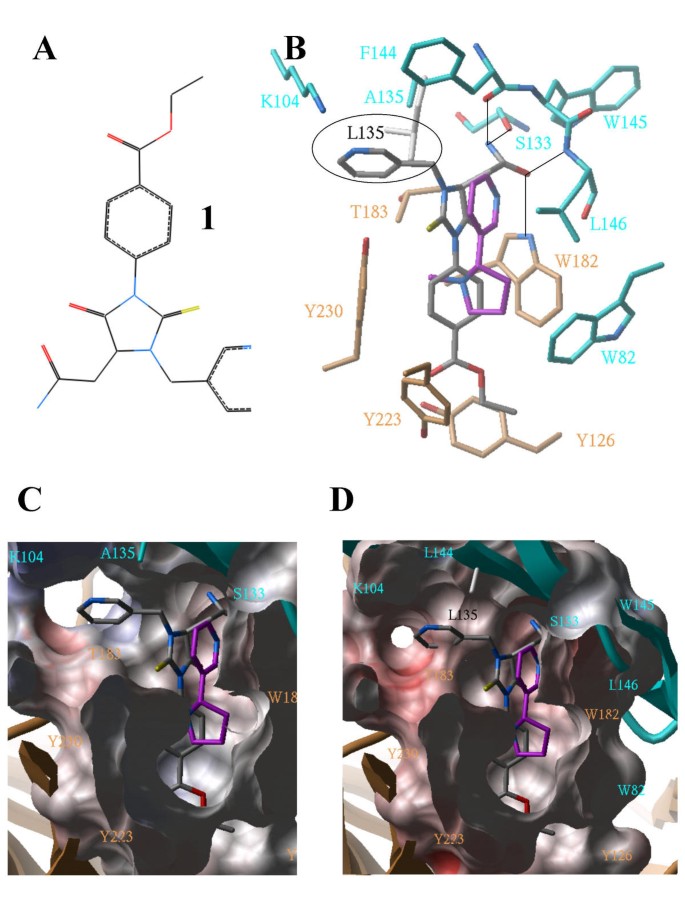
Candidate nAChR agonist, specific for beta2-containing subtypes. A: Chemical structure of 1. B: Details of the interactions between 1 (gray) and alpha4-beta2 nAChR agonist binding pocket (gold: alpha4, cyan: beta2). 1 occupies the same pocket as docked nicotine (magenta), and presents a pyridine extension (circled in black) that fits next to Ala 135 of beta-2, but clashes with Leu 135 of beta-4 (white), or Val 135 of alpha-7 (not shown). An amide group of 1 fills the pocket occupied by a water molecule when nicotine or acetylcholine are bound (Figure 3 – Left) and makes a network of hydrogen-bonds (black lines) with the receptor.C: Same view and orientation as in B, with a skin representation of the binding pocket.D: Same as C, with alpha4-beta4 nAChR binding pocket: the steric clash between the receptor and the pyridine moiety of the ligand is visible.

To address further the specificity of this ligand candidate, we docked it, as previously described, to the other three nAChRs subtypes studied in this work, as well as to the crystal structure of AChBP, and calculated the corresponding binding energies (Table 1 bottom). In all cases, the predicted binding energies are better than for nicotine or acetylcholine. The calculated Van der Waals repulsions with alpha4-beta2 and alpha3-beta2 nAChRs remain small, and within the accuracy limit of Van der Waals energy prediction (the otherwise negligible displacement of an atom by a tenth of an Angstrom may result in several kcal/mol energy difference). On the other hand, the very high Van der Waals repulsions calculated Table 1 (bottom) for alpha4-beta4 and alpha7-alpha7 are a clear indication of a steric clash (Figure 5D): the ligand fits next to Ala 135 of beta2, but cannot accommodate Leu 135 of beta4.
Conclusions
Previous reports have shown on other systems that a single-residue mutation in a receptor is sufficient for the development of specific ligands [13, 14]. Based on the recent crystal structure of AChBP, we could propose a detailed description of the structural variations of nAChR subtypes. Our models of the nAChRs binding pocket are only predictive, and need to be confirmed experimentally; testing in vitro the activity of ligand candidates, such as 1, derived from the structure of the receptor would constitute an important first step towards the validation of our models as tools for the discovery of novel nAChR agonists.
Materials and Methods
Multiple Sequence Alignment
The sequences of AChBP, and of the alpha3, alpha4, alpha7, beta2 and beta4 nAChR monomers were aligned using the ICM multiple sequence alignment algorithm that includes the following steps [16, 10]: 1) pairwise alignment using Needleman and Wunsch algorithm modified to use zero end gap penalties (so called ZEGA) [15, 16]. 2) evolutionary tree construction with a "neighbor-joining algorithm" to determine the order of alignment and calculate relative weights of sequences and profiles from the branch lengths [17]. 3) traverse the tree from top to bottom, aligning the closest sequences or profiles.
3D Model Building of nAChRs Subunit Interface
For each nAChR dimer, two monomer sequences were threaded onto two consecutive subunits of the AChBP crystal structure, based on the multiple sequence alignment, and the energy of the system was locally minimized by the ICM method [10, 18]. The orientation of all polar hydrogens was systematically optimized through conformational space search combined with local minimization. All nAChR residues within 7A of the putative AChBP binding pocket described by Brejc et al. [6] that differed from the AChBP template residue were energy-minimized in the internal coordinates space using ICM global conformational search algorithm. Energy terms included Van der Waals, electrostatic, hydrogen bond, and torsion terms [10].
Receptor-Ligand Docking
The flexible ligand / grid receptor docking algorithm implemented in ICM was applied, with ECEPP/3 force field and MMFF partial charges for the ligand, and hydrophobic, electrostatic, hydrogen-bond and surface potential maps for the receptor [18, 19, 10]. Further optimization with flexible ligand and full atom representation of the receptor was carried out by local energy minimization, where all side-chains within 6 A of the ligand were flexible [18]. When docking was performed in the presence of a water molecule, the later was included in the grid representation of the receptor, and was mobile during full-atom local minimization.
Binding Energy Calculation
The binding energy function implemented in icm is partitioned into three terms: the hydrophobic, or cavity term, that accounts for the variation of water/non-water interface area, the electrostatic term, composed of the coulombic interactions and desolvation of partial charges calculated by the boundary elements method, and the entropic term, that results from the decrease in the conformational freedom of functional groups buried upon complexation [20]. A constant term also present in the energy function, that accounts for the change of entropy of the system due to the decrease of free molecules concentration and the loss of rotational/translational degrees of freedom, was omitted, and only differences in the binding energies of the ligands are accurate. The dielectric constant and surface tension utilized were optimized against a benchmark of eight protein-protein, 13 protein-small ligand, and eight protein-peptide complexes, and result in a 2.5 kcal/mol accuracy [20].
Virtual Library Screening
The high-throughput docking implemented in the ICM program [10] was applied. The procedure followed for each ligand was similar than the grid docking previously described [21]: each flexible ligand of the ChemDiv public library (ChemDiv Inc., San Diego) was docked automatically into the combination of potential maps described above, and assigned a score according to its fit with the alpha4-beta2 nAChR. The scoring function included continuum as well as discreet electrostatics, hydrophobicity and entropy parameters. The screening of the database of over 200,000 ligands took less than 2 days on a Linux cluster of 150 processors. The 3003 compounds which scored better (i.e. lower) than -35 were pre-selected for a second automatic round of selection: only ligands within 5 A of Ala 135 of the beta-2 subunit and that were hydrogen-bonded to the carbonyl oxygen of beta-2 Ser 133 were retained. 1 was among the best scoring such compounds after this second round of selection.
References
Goodman Gilman A: The Pharmacological Basis of Therapeutics. Ninth Edition, 1995, 177–197.
Changeux JP, Bertrand D, Corringer PJ, Dehaene S, Edelstein S, Lena C, Le Novere N, Marubio L, Picciotto M, Zoli M: Brain nicotinic receptors: structure and regulation, role in learning and reinforcement. Brain Res. Rev. 1998, 26: 198–216. 10.1016/S0165-0173(97)00040-4
Holladay MW, Dart MJ, Lynch JK: Neuronal nicotinic acetylcholine receptors as targets for drug discovery. J. Med. Chem. 1997, 40: 4169–94. 10.1021/jm970377o
Lloyd GK, Williams M: Neuronal nicotinic acetylcholine receptors as novel drug targets. J. Pharmacol. Exp. Ther. 2000, 292: 461–7.
Miyazawa A, Fujiyoshi Y, Stowell M, Unwin N: Nicotinic acetylcholine receptor at 4.6 A resolution: transverse tunnels in the channel wall. J. Mol. Biol. 1999, 288: 765–86. 10.1006/jmbi.1999.2721
Brejc K, van Dijk WJ, Klaassen RV, Schuurmans M, van Der Oost J, Smit AB, Sixma TK: Crystal structure of an ACh-binding protein reveals the ligand-binding domain of nicotinic receptors. Nature 2001, 411: 269–76. 10.1038/35077011
Kao PN, Karlin A: Acetylcholine receptor binding site contains a disulfide crosslink between adjacent half-cystinyl residues. J. Biol. Chem. 1986, 261: 8085–8.
Abagyan R, Totrov M: Biased probability Monte Carlo conformational searches and electrostatic calculations for peptides and proteins. J. Mol. Biol. 1994, 235: 983–1002. 10.1006/jmbi.1994.1052
Zhong W, Gallivan JP, Zhang Y, Li L, Lester HA, Dougherty DA: From ab initio quantum mechanics to molecular neurobiology: a cation-pi binding site in the nicotinic receptor. Proc. Natl. Acad. Sci. USA 1998, 95: 12088–93. 10.1073/pnas.95.21.12088
Molsoft LLC: ICM2.8 manual.1999. [http://www.molsoft.com]
Grutter T, Changeux JP: Nicotinic receptors in wonderland. Trends Biochem. Sci. 2001, 26: 459–463. 10.1016/S0968-0004(01)01921-1
Grutter T, Ehret-Sabatier L, Kotzyba-Hibert F, Goeldner M: Photoaffinity labeling of Torpedo nicotinic receptor with the agonist [3H]DCTA: identification of amino acid residues which contribute to the binding of the ester moiety of acetylcholine. Biochemistry 2000, 39: 3034–43. 10.1021/bi992393o
Mohammadi M, McMahon G, Sun L, Tang C, Hirth P, Yeh BK, Hubbard SR, Schlessinger J: Structures of the tyrosine kinase domain of fibroblast growth factor receptor in complex with inhibitors. Science 1997, 276: 955–60. 10.1126/science.276.5314.955
Klaholz BP, Mitschler A, Moras D: Structural basis for isotype selectivity of the human retinoic acid nuclear receptor. J. Mol. Biol. 2000, 302: 155–70. 10.1006/jmbi.2000.4032
Needleman SB, Wunsch CD: A general method applicable to the search for similarities in the amino acid sequence of two proteins. J. Mol. Biol. 1970, 48: 443–453.
Abagyan RA, Batalov S: Do aligned sequences share the same fold? J. Mol. Biol. 1997, 273: 355–368. 10.1006/jmbi.1997.1287
Saitou N, Nei M: The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol 1987, 4: 406–425.
Totrov M, Abagyan RA: Flexible protein-ligand docking by global energy optimization in internal coordinates. Proteins Suppl. 1997, suppl 1: 215–220.
Totrov M, Abagyan RA: in Drug-Receptor Thermodynamics: Introduction and Application. John Wiley & Sons, Ltd 2001, 603–24.
Schapira M, Totrov M, Abagyan R: Prediction of the binding energy for small molecules, peptides and proteins. J Mol Recognit. 1999, 12: 177–90. 10.1002/(SICI)1099-1352(199905/06)12:3<177::AID-JMR451>3.3.CO;2-Q
Schapira M, Raaka B, Samuels HH, Abagyan R: Rational discovery of nuclear receptor antagonists. Proc. Natl. Acad. Sci. USA 2000, 97: 1008–1013. 10.1073/pnas.97.3.1008
Acknowledgements
This work was partly supported by STTR grant 1 R41 DK59041-01 from the NIH, and SBIR grant 65559S01-I from the DOE awarded to Molsoft.
Author information
Authors and Affiliations
Corresponding author
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
Rights and permissions
About this article
Cite this article
Schapira, M., Abagyan, R. & Totrov, M. Structural model of nicotinic acetylcholine receptor isotypes bound to acetylcholine and nicotine. BMC Struct Biol 2, 1 (2002). https://doi.org/10.1186/1472-6807-2-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1472-6807-2-1